

Your Kitchen Is a Pharmacy.
Here's the Proof.

By Steven Muskal, Ph.D. · CEO, Eidogen-Sertanty, Inc. · June 1, 2026 | stevenmuskal.com
“The most powerful medicines aren’t locked away in pharmaceutical patents. They’re sitting on your dinner table, and we can prove it, molecule by molecule.”
Let me give you a number: 0.98 out of 1.0.
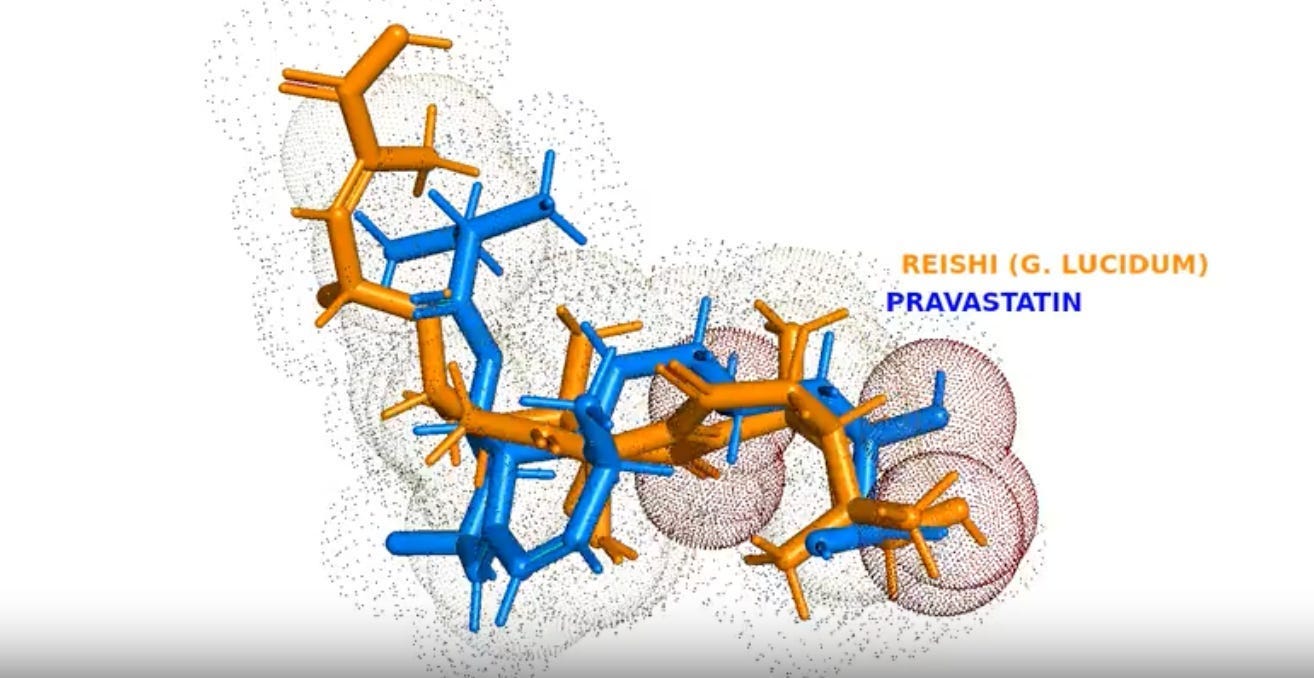
That’s the pharmacophore similarity score between Pravastatin - a blockbuster cholesterol drug that generated billions of dollars in annual revenue during its peak years - and Ganoderic acid, a compound found in Reishi mushrooms that cost-per-gram less than a cheap cup of coffee. Nearly identical. In three-dimensional molecular space, these two substances - one born in a pharmaceutical lab, one evolved in a forest - present almost exactly the same biological “face” to the enzymes that regulate your cholesterol.
This isn’t folklore. This isn’t “wellness influencer” speculation. This is the output of pharmaceutical-grade computational chemistry - the same class of methods that created multi-billion-dollar drug pipelines - pointed back at nature. This is Drug to Table.
The Radical Proposition
Indeed, the most powerful medicines might not be locked in pharmaceutical patents. They might be sitting on your dinner table.
For decades, drug companies have used sophisticated computational tools to design synthetic molecules that interact with human biology. These methods have created life-saving medications - but they’ve remained trapped behind corporate walls, optimized for patent protection rather than public health.
We can change that. By turning these same tools toward nature, we can identify therapeutic molecules in foods, spices, and traditional remedies - compounds that already exist and can’t be monopolized.
I call this “Drug to Table”: mining nature’s pharmacy with pharmaceutical-grade intelligence.
“Nature has been running the ultimate drug discovery program for 400 million years - and the results are sitting in your spice cabinet.”
The Core Idea: For 50 years, pharmaceutical companies used sophisticated computational methods to mine nature’s molecular library - then patented what they found. Drug to Table reverses the flow: we take the drugs that already exist, reverse-engineer their biological “keys,” and find those same keys hidden in foods, mushrooms, and spices you can buy at any grocery store.
How Nature Beat Big Pharma - By 400 Million Years
The Patent Problem
Pharmaceutical companies operate under a fundamental commercial constraint: they can only profit from molecules that are novel enough to patent, synthetic enough to control manufacturing of, and complex enough to prevent generics from undercutting them. This creates a perverse incentive structure: ignore nature’s existing solutions in favor of patentable synthetic alternatives. Pharma also faces the innovator’s dilemma, where pursuing disruptive new approaches can threaten existing revenue streams. Together, these forces may reduce incentives to pursue compounds that could significantly impact health while disrupting established franchises.
Nature doesn’t care about patents or cannibalizing past approaches. It is truly opportunistic and continually seeking improved overall fitness. Plants have been running the ultimate drug discovery program for 400 million years, evolving molecules to defend against pathogens, attract beneficial organisms, survive environmental stress, and compete with neighboring species. The result is a molecular library of unimaginable diversity - all battle-tested by evolution, all freely available to anyone.
What Pharmaceutical Companies Actually Did
Here’s the irony they don’t advertise: the first statins (lovastatin, simvastatin) weren’t invented - they were discovered in fungi. Aspirin came from willow bark. Penicillin from mold. Taxol, one of the most effective cancer drugs ever made, comes from the Pacific yew tree. The entire golden age of pharmaceutical development was built on mining nature’s pharmacy.
Then they stopped looking - because they’d patented enough to be profitable. The question Drug to Table asks is: what other easy-to-access, less expensive solutions are out there?
The Computational Revolution: Beyond Guesswork
Traditional approaches to finding natural medicines rely on folklore, basic chemical similarity, or sheer luck. Modern pharmaceutical discovery uses sophisticated methods - and that same precision is now available to open science:
QSAR Modeling: Quantitative Structure-Activity Relationships predict how strongly molecules will interact with specific biological targets - the protein “locks” that drugs are designed to engage.
Molecular Fingerprinting: Instead of matching flat chemical drawings, this captures how molecules present themselves in 3D space - their actual functional shape, the geometry that determines biological activity.
These methods can find “non-obvious functional equivalents” - natural compounds that work like known drugs but look completely different on paper. That’s the key insight that changes everything.
Understanding Pharmacophores: The Molecular Lock-and-Key System
At the heart of Drug-to-Table discovery lies the concept of pharmacophores - the specific 3D arrangements of chemical features that allow molecules to interact with biological targets. Think of them as molecular keys. The lock is a protein receptor. And two completely different-looking keys can open the same lock - if their functional teeth are arranged identically.
A pharmacophore is defined by three elements:
Chemical features: What types of interactions can occur (hydrogen bonding, hydrophobic contact, charge interactions)
Spatial arrangement: Where these features are positioned in 3D space
Distance relationships: How far apart these features are from each other
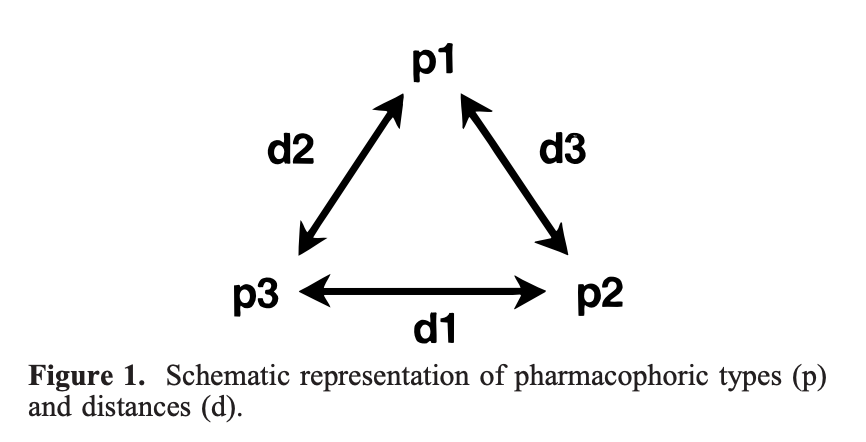
Fig. 1 — The PharmPrint pharmacophore basis set: 10,549 three-point pharmacophore descriptors encoding molecular interaction patterns in 3D space. From McGregor & Muskal (1999).
Pharmacophore Fingerprinting: The Science of Molecular “Keys”
Think of every drug as a key, and every biological target (enzyme, receptor, protein) as a lock. The drug doesn’t just need to be vaguely the right size - it needs to present specific chemical features in specific three-dimensional positions: hydrogen bond acceptors here, hydrophobic carbon chains there, charged regions at exact spatial intervals.
Pharmacophore fingerprinting captures this 3D “keycode” as a set of mathematical descriptors - a compact representation of how a molecule will behave in biological space, regardless of what it looks like on paper.
My decades-old PharmPrint work (published as a series in Journal of Chemical Information and Computer Sciences, 1999: Pharmacophore Fingerprinting. 1. Application to QSAR and Focused Library Design AND Pharmacophore Fingerprinting. 2. Application to Primary Library Design) used a basis set of 10,549 three-point pharmacophores, constructed by enumerating different pharmacophoric feature types — Acceptor, Donor, Hydrophobic, Negative charge, Positive charge, and aRomatic — across various distance ranges. This creates a comprehensive “vocabulary” for describing molecular interactions at the 3D functional level.
Fig. 2 (Fig. 4 in the original publication) Multi-conformer pharmacophore comparison: each molecular shape generates an independent fingerprint, enabling precise 3D alignment matching.
QSAR Modeling: Predicting Potency Before You Touch a Test Tube
Quantitative Structure-Activity Relationships (QSAR) can use these fingerprints to predict how strongly a candidate molecule will interact with a biological target. Combined with pharmacophore screening, this means we can rank thousands of food compounds by predicted biological activity - before a single experiment is run.
In our 1999 series, we illustrated that full drug-target classes can be resolved into separable feature space when representing molecules with PharmPrints.
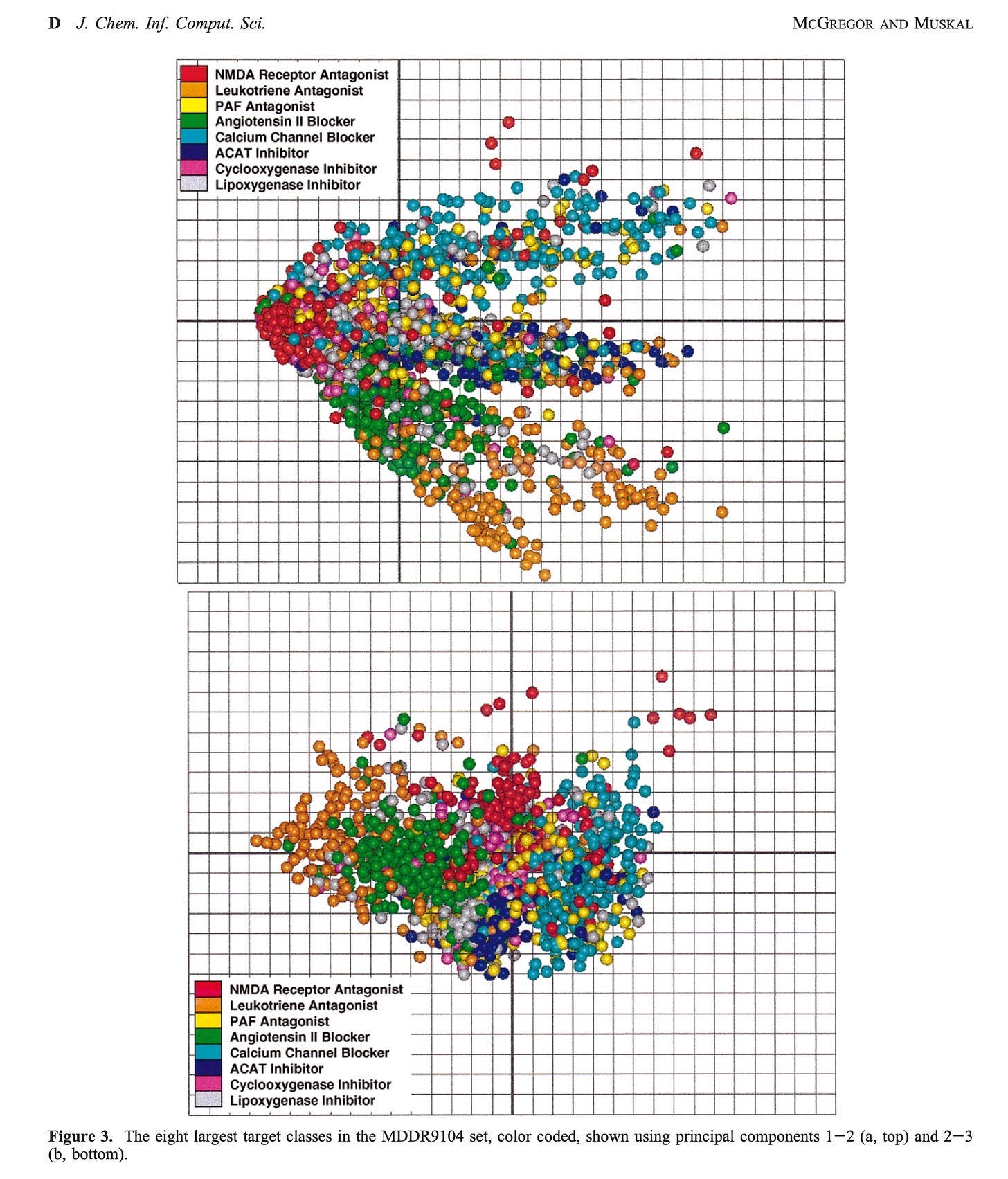
Fig. 3 Major drug-family classes can be clustered using PharmPrint representations of chemical structure. Molecules with the same or similar PharmPrints, often with very different 2D structures, can be mapped into the same space. PharmPrints can thus leverage the concept “similar molecule → similar activity.”
Non-obvious Me-Too’s
Years ago, with PharmPrints we illustrated two molecules which look very different from a two-dimensional perspective (e.g. the kind patent examiners look at) can share very similar pharmacophoric feature, that is, PharmPrint similarity (PharmSim). Below is one example. Zantac and Tagamet were blockbuster drugs one from Glaxo (Zantac) and another from SmithKline Beecham (Tagamet). Each held separate patent positions and each drug generated enormous revenue for both organizations. In the year 2000, these organizations merged into what is now called GSK. Zantac ultimately crushed Tagamet in revenue, generating an estimated $25–30 billion over its lifetime compared to Tagamet’s estimated lifetime revenue of $14 billion.

The Evolution: From Single Fingerprints to Multi-Conformer Analysis
The Original PharmPrint Method (published 1999) created a single binary fingerprint per molecule by combining all possible pharmacophore patterns into one compact descriptor - imagine compressing all of a molecule’s 3D interaction possibilities into a single barcode for rapid comparison against thousands of others. Excellent for screening scale.
Of course, molecules aren’t rigid. In solution, they twist and flex, adopting dozens or hundreds of different three-dimensional shapes (conformers) - each of which can interact differently with biological targets. My newer PolyPharmPrint technology generates individual pharmacophore fingerprints for each conformer, then compares entire ensembles of shapes between molecules.
This is the difference between saying “these people have similar faces” and being able to specify exactly which facial features match at sub-millimeter precision. From coarse similarity to exact molecular alignment.
Instead of one fingerprint per molecule, we now generate:
Multiple conformers for each molecule (representing different 3D shapes a molecule may adopt in solution)
Individual fingerprints for each conformer
Ensemble comparisons across all possible shapes
When we find a match between conformers, we can visualize the exact 3D alignment that explains the biological similarity. This is no longer “similar-ish” - this is molecular-level proof.
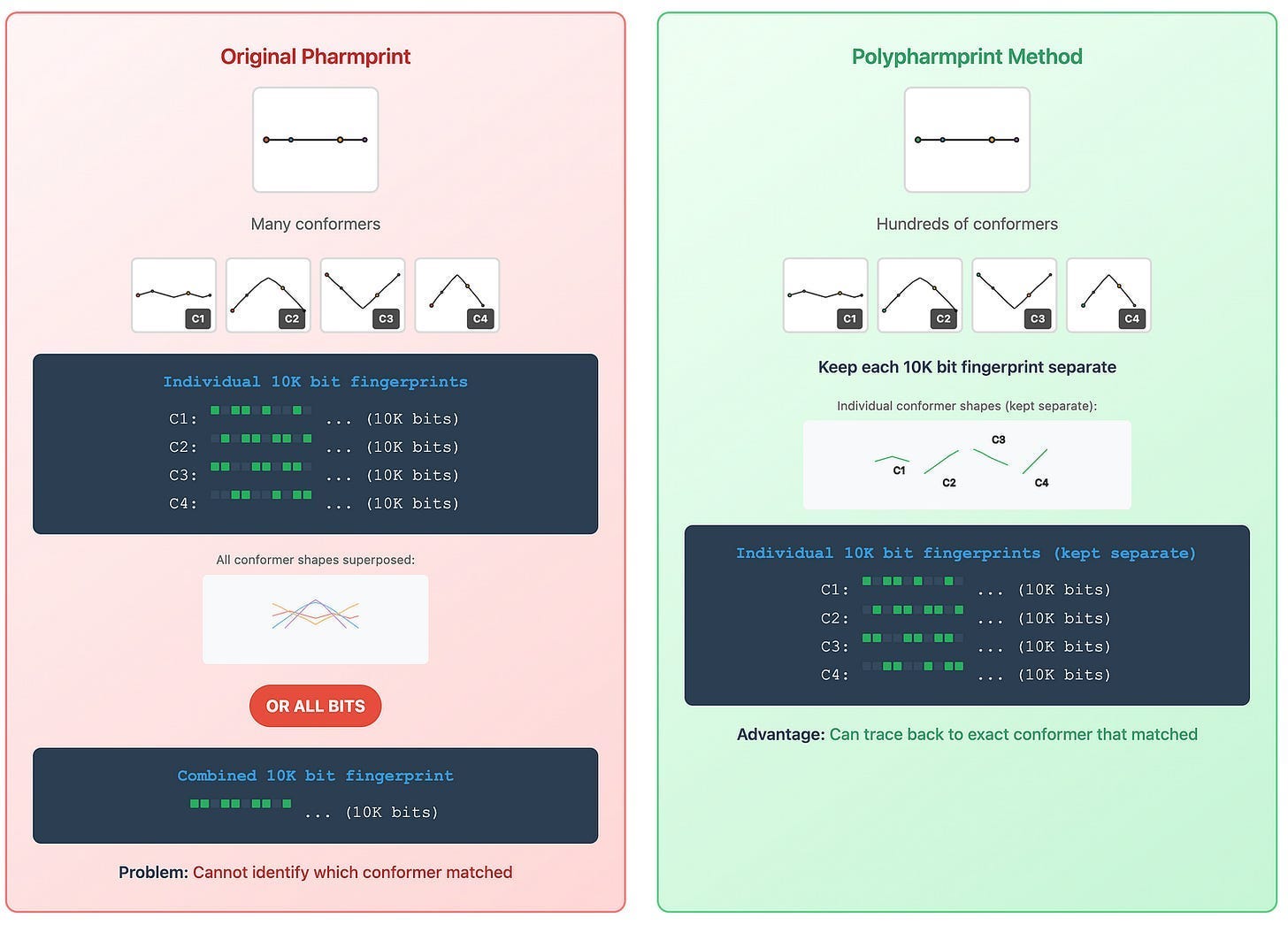
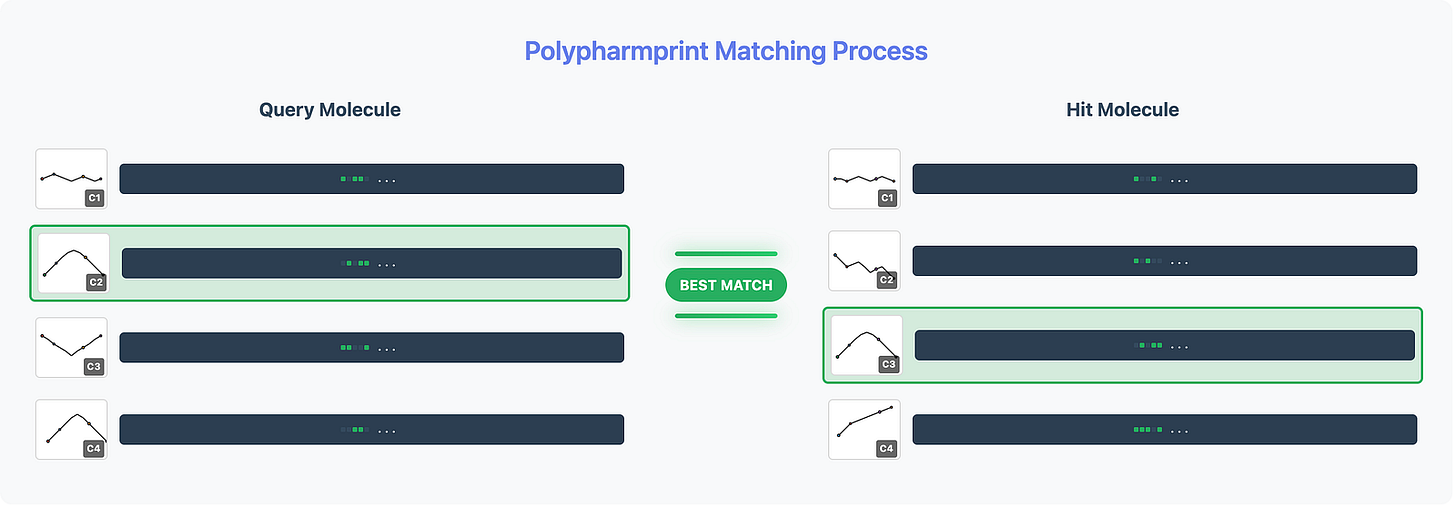
Fig. 4 The evolution of PharmPrints into PolyPharmPrints. The advance from single to ensemble fingerprinting dramatically improves match specificity.
Key Breakthrough:
We can access the exact 3D conformation of the matching conformer PharmPrint, enabling dramatically more efficient exploration of the conformational space when aligning query and hit molecules. This transforms molecular alignment from a computationally expensive global search into a targeted, efficient process. It also affords the opportunity for rapid and easy visual inspection confirming molecular superposition(s).
Case Study: The Mushroom That Thinks It’s a Statin
How computational methods revealed a hidden connection between a billion-dollar drug and an ancient mushroom used medicinally for 2,000 years.
The Starting Point
Pravastatin is a blockbuster statin drug - one of the most prescribed medications on earth. It works by blocking HMG-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis. Hundreds of millions of people take statins. Billions of dollars flowed from them annually.
Here’s the twist: the original statins weren’t invented in a lab. Lovastatin, the first in the class, was isolated from Aspergillus terreus - a fungus. Nature evolved HMG-CoA reductase inhibitors in fungi millions of years ago. We just found them and patented the synthetic derivatives.
This raised a question I couldn’t shake: if nature evolved cholesterol-lowering compounds once, where else might they exist?
The Computational Hunt
Using PolyPharmPrint screening, I analyzed Pravastatin’s 3D molecular fingerprint across its full conformer ensemble and ran it against the 28,000+ food chemicals in the FooDB database. The goal wasn’t to find something that looked like Pravastatin on paper - it was to find something that would function like Pravastatin in the body.
The Stunning Discovery
Ganoderic acid A from Reishi mushrooms (Ganoderma lucidum) emerged as a near-perfect functional match.

See the Alignment
360° rotating visualization of the 3D structural alignment between Pravastatin (blue/cyan) and Ganoderic acid from Reishi mushrooms (orange/red). The molecules have been optimally superimposed using PolyPharmpPrint technology. Explore the full interactive Drug-to-Table visualization →
What the Alignment Means
On paper, Pravastatin and Ganoderic acid A look different. But in three-dimensional biological space - where interactions with enzymes actually happen - they present nearly identical patterns:
Hydrogen bond acceptors (the red oxygen atoms in the video) sit in matching positions
Hydrophobic carbon chains that repel water align identically, helping both molecules anchor in the same enzyme binding pockets
Overall 3D shape occupies the same space — like finding two completely different keys that open the same lock because they have identical functional teeth
Ancient Wisdom, Modern Validation
Reishi has been called the “mushroom of immortality” in Chinese medicine for over 2,000 years, specifically recommended for cardiovascular health and “blood stasis.” Published studies report that Reishi extracts may reduce total cholesterol, LDL, and triglycerides in both animal and human studies. The mechanism? Consistent with HMG-CoA reductase inhibition - exactly what our 3D analysis predicted.
The traditional healers didn’t have molecular modeling software. They had something different: thousands of years of careful observation. Drug to Table’s computational methods provide the molecular explanation for what they knew empirically.
The Validation Trail:
Computational: 0.98/1.0 pharmacophore similarity via PolyPharmPrint multi-conformer analysis
Traditional: 2,000+ years of documented cardiovascular use in TCM
Laboratory: Multiple peer-reviewed studies confirming cholesterol reduction in human trials
Personal: Ongoing self-experiment - standardized Reishi extract with regular lipid panel monitoring
→ Explore the full technical analysis, 3D visualization, and PolyPharmPrint methodology
The Broader Implication
If this level of molecular similarity exists between a top-selling drug and a common mushroom, how many more pharmaceutical-food connections are waiting to be discovered among the other 28,000 food chemicals - or the 400,000+ natural products in the COCONUT database?
This is why we built the tools to bring this capability to everyone. Starting with your phone.
Food Is Medicine in Your Pocket
Computational science tells us which foods have drug-like properties. But knowing that Reishi contains statins doesn’t help you if you can’t track whether you’re actually eating functional foods, in functional amounts, with measurable results. That’s the gap we built an app to close.



Food Health Screen
Your daily bridge between food science and your plate.
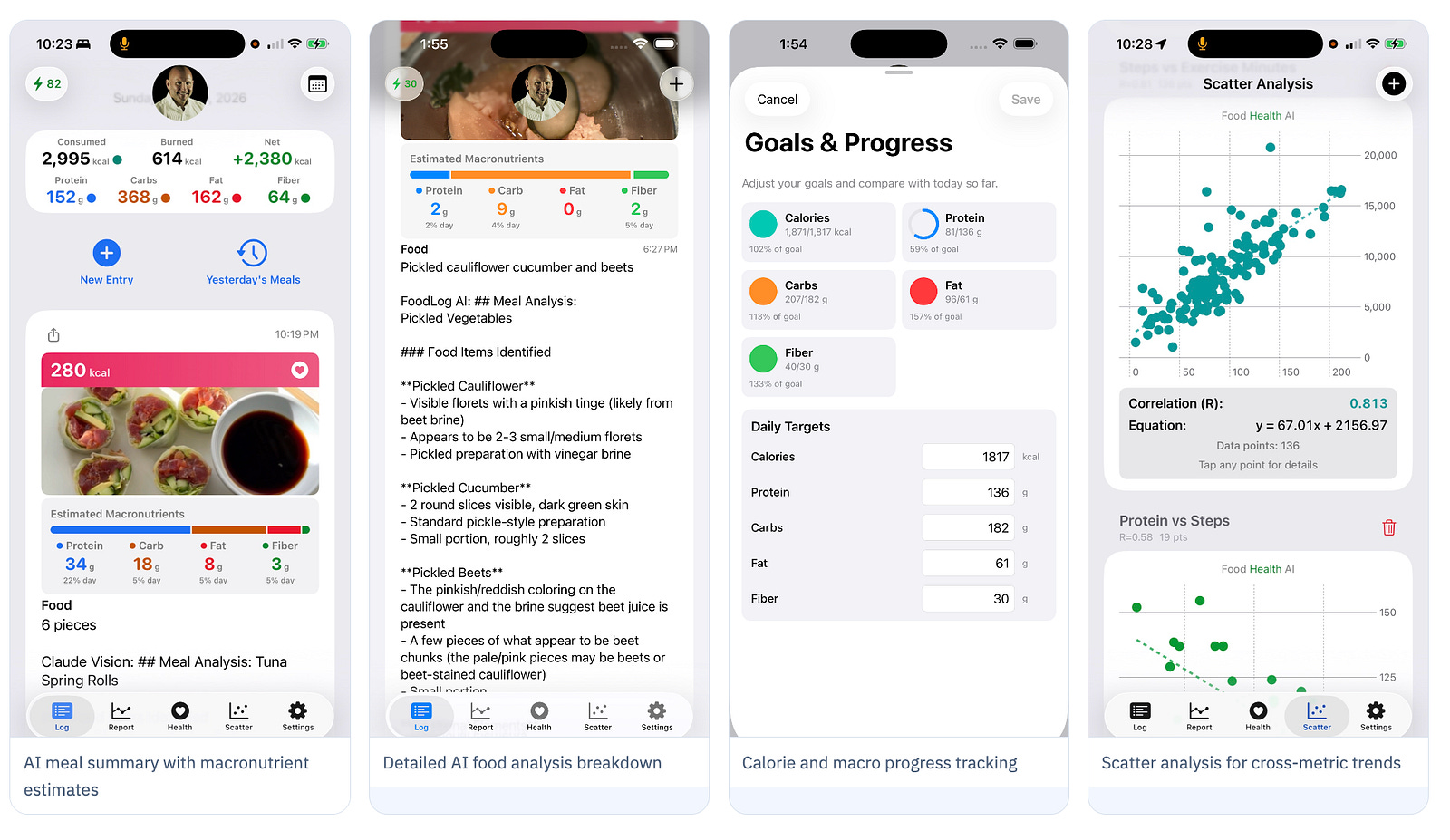
Food Health is the consumer-facing application of exactly the philosophy described in this article. Point your phone at any meal - a bowl of Reishi broth, a turmeric-spiced dish, a restaurant menu - and get instant AI-powered nutritional analysis: calories, protein, carbs, fat, and fiber in seconds. This is the first step.
But Food Health goes beyond calorie counting. Its scatter analysis and trend reporting let you do something that was previously only possible in research settings: correlate what you eat with how you feel, sleep, and perform - over days, weeks, and months. This is citizen science applied to your personal biology. I see this as a foundational first step to a new approach to natural product screening where anyone with a phone and a camera can participate
📸 Photo & Note Analysis AI estimates calories and macros from a photo or a typed description in seconds.
❤️ Apple Health (iOS) / Health Connect (Android) Sync See nutrition alongside sleep, activity, and heart rate — the full picture.
📊 Scatter Analysis & Reports Discover personal correlations: does turmeric improve your HRV? Does Reishi affect your sleep?
🔒 Local-First Privacy Your data stays on your device by default. You control everything.
📱Log Now, Analyze Later Capture meals even offline. Run AI analysis when you’re connected.
⌚Device Integration Custom prompts for glucose meters, blood pressure cuffs, and more.
Food Health TxD
Carbs & glucose for diabetes — spot the meals that spike you


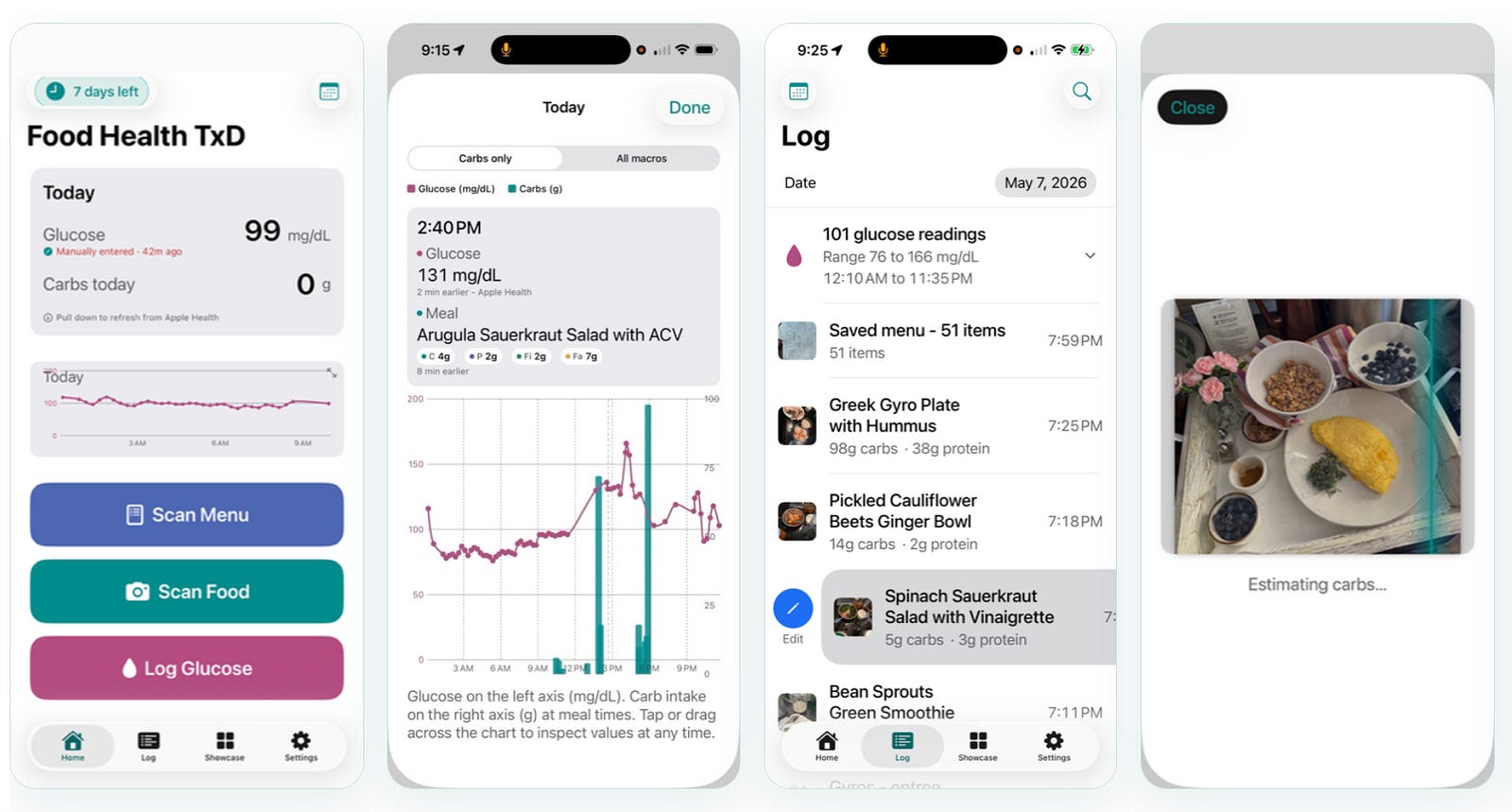
For the 130 million Americans with diabetes or prediabetes, the drug-to-table insight is immediately urgent: every meal is a metabolic event. Food Health TxD brings AI meal scanning together with continuous glucose monitoring (CGM) data — so you can see, in real time, exactly how each food affects your blood sugar.
📸 AI meal scanning Instant carb and macro estimates from a photo
📉 Glucose overlay CGM readings and meals on the same timeline
📋 Menu scanner Scan a restaurant menu, pick the lowest-carb options
❤️ Apple Health (iOS) / Health Connect (Android) sync Reads glucose, writes carbs automatically
📊 Trend analysis Spot patterns between meals and spikes over weeks
As I wrote in May 2026: “Half of American adults are prediabetic or diabetic. This isn’t a headline - it’s a quiet emergency happening in every household.” TxD was built to give people a simple way to see how food affects their glucose, one meal at a time.
Food Health TxD is for education and tracking only. It is not a medical device and does not provide diagnoses, insulin dosing recommendations, or clinical predictions. Consult your healthcare provider before making changes to your treatment plan.
Your Personal Drug Discovery Lab
The computational pipeline that produced the Pravastatin-Reishi discovery is not locked in a university basement. We may just integrate the pipeline into the Food Health scan in Food Health TxD apps in future versions. But if you are interested in going at it yourself - with open-source tools and public databases, you can run versions of this same analysis - or, at the very least, use its outputs to make more informed daily food choices.
Target Selection
Choose a biological pathway you care about: cholesterol metabolism, blood sugar regulation, inflammation, cognitive function, sleep architecture. Pick a known drug that addresses it - that’s your computational starting point.
Computational Screening
Generate pharmacophore fingerprints from the drug. Several open source pharmacophoric generation utilities are available. Screen natural product databases (FooDB’s 28,000+ food chemicals; COCONUT’s 400,000+ natural products; ChEMBL’s biological activity data). Apply QSAR predictions to rank candidates by predicted activity. Cross-reference with traditional use and safety literature.
Systematic Exploration
Source the top candidates - ideally standardized extracts with known compound concentrations. Design a personal protocol: baseline biomarker measurement, defined supplementation period, repeat measurement. Track everything with Food Health Scan.
Data, Not Anecdote
Use Food Health’s scatter analysis to correlate your intake with objective metrics: sleep quality, heart rate variability, energy levels, blood glucose (with TxD if relevant). This turns personal experimentation into structured self-science.
The Spice Cabinet Pharmacy
The Pravastatin-Reishi case isn’t an isolated curiosity. Drug to Table screening of the top 200 pharmaceuticals is ongoing, and provisional findings suggest the spice cabinet is far more medicinally dense than most people realize:
Turmeric (Curcumin): Computational analysis reveals similarity to multiple COX-2 anti-inflammatory drugs — the same target as ibuprofen and celecoxib, without the gastrointestinal side effects at culinary doses.
Garlic (Allicin): Pharmacophore patterns consistent with ACE inhibitors used for blood pressure management. Traditional cardiovascular use across dozens of cultures suddenly makes molecular sense.
Ginger (Gingerols): Structural relationships to 5-HT₃ antagonists — the class of drugs used clinically for nausea. Why does ginger settle an upset stomach? Now we can show you exactly why.
Cinnamon (Cinnamaldehyde): Computational similarity to insulin sensitizers — consistent with traditional use of cinnamon to moderate blood sugar. Food Health TxD users: this is one worth tracking.
Reishi (Ganoderic acids): Our flagship discovery — near-perfect pharmacophoric overlap with Pravastatin. The “mushroom of immortality” earns its name computationally.
Traditional Medicines Examples
Computational methods are now providing the molecular explanations for what traditional healers discovered empirically over millennia:
Willow Bark → Aspirin: The original pharmacological natural product -salicylate’s mechanism is now textbook biochemistry
Foxglove → Digitalis: Computational analysis may explain why this traditional heart remedy modulates cardiac contractility
Berberine (Goldenseal, Barberry): Shows metformin-like computational profiles for glucose regulation - of strong interest to Food Health TxD users
Reishi → Statin-like effects: Our current discovery validates and molecularly explains 2,000 years of cardiovascular use
Important context: Computational similarity does not equal clinical equivalence. Bioavailability, metabolism, dose, and individual genetics all matter enormously. These findings suggest promising directions for personal investigation - not prescriptions. Always consult your healthcare provider about any significant health concern.
💡 The Network Effect: Community-Driven Validation
As more people adopt these methods and use tools like Food Health to track their nutritional intake and health correlations, we build collective intelligence:
Shared protocols for systematic personal investigation
Open databases of personal results and meal-to-metric correlations
Collaborative refinement of computational models
Crowdsourced identification of effective natural approaches — validated by real human data, not just algorithms
The Democratization Opportunity
“For the first time in history, the same computational intelligence used to discover billion-dollar drugs is available to anyone - applied to natural compounds that belong to everyone.”
This isn’t about replacing pharmaceutical innovation. It’s about expanding the toolkit available for understanding how food choices might influence biological systems. And critically, it’s about closing the gap between discovery and action.
The Drug-to-Table computational methodology identifies candidates. Food Health makes the daily practice of food-as-medicine measurable. Together, they represent a new paradigm: molecular intelligence meets personal health data.
For the first time in history, the computational intelligence used to discover billion-dollar drugs is accessible to individuals. Not the full pharmaceutical apparatus - but the core intellectual methods: pharmacophore fingerprinting, QSAR modeling, open molecular databases.
And now, the consumer interface for living those discoveries is in your pocket. Food Health AI closes the loop between:
💡 Computational insight (Drug to Table methodology)
🍽️ Daily food choices (what you actually eat)
📊 Measurable outcomes (biomarkers, trends, correlations)
This is not Big Pharma. This is your pharmacy. The same computational power that generated patent portfolios worth billions can now help you make better decisions at the grocery store, at the spice market, and at your kitchen table.
“We’re not discovering that foods contain drug-like molecules. We’re remembering that drugs came from food in the first place - and building the tools to act on that knowledge.”
What’s Next
The Pravastatin-Reishi connection is one data point in what will become a publicly accessible atlas of drug-food molecular relationships. Here’s what’s coming:
📡 Complete computational protocols for Drug-to-Table discovery
🍄 Results from screening the top 200 pharmaceuticals against 28,000+ food chemicals
🧪 Personal testing frameworks for systematic self-experimentation (with Food Health AI integration)
🌐 Community database for sharing computational hits, personal protocols, and biomarker results
📱 Planned Food Health AI features: functional food tagging, compound tracking, drug-food interaction alerts
All protocols, databases, and methodologies will be published as open resources. The goal is to make molecular nutrition intelligence accessible to anyone curious enough to use it.
Follow along:
📱 Food Health AI (iOS & Android)
📱 Food Health TxD (iOS)
⚠️ Important Disclaimer
This article represents computational research methodology for educational and wellness exploration purposes only. It is not medical advice.
Any health concerns should be addressed with qualified healthcare professionals. Natural compounds can have potent biological effects and potential interactions with medications — particularly important if you are currently taking statins or other prescription medications.
Correlational findings: Pharmacophore similarity does not guarantee identical biological activity. Actual effects depend on bioavailability, metabolism, dosage, individual genetics, and physiology. Structural similarity is a hypothesis-generating tool, not a clinical proof.
Natural ≠ Safe: Many plants contain potent bioactive compounds. Always consult a healthcare provider before using supplements alongside prescription medications.
Food Health and Food Health TxD are consumer wellness tracking applications, not medical devices. They do not provide diagnoses, clinical predictions, or treatment recommendations.

Steven Muskal, Ph.D. is the CEO of Eidogen-Sertanty, Inc. — a drug discovery informatics company. He has spent four decades working at the intersection of computational biology, AI, and drug discovery. He writes about AI, health, and the intersection of biology and technology at stevenmuskal.com
For a couple mixes, I wanted to pull teo from the archives. The first one was fun, especially if you know the back story. TimD vocals/guitar, Andrew Guitar, Rick Vocals, AlanW Bass, and Johnny Conga. The Way (first and only pass):
Then during a July 3rd mix (as we start planning for one this year) - The Story with neighbors. Maya leading vocals, Tammy and Pam assisting vocals, Dom Guitar, Jesse Guitar, Don Keys, TimH Bass:
I have another example for the “Spice Cabinet Pharmacy” list. This one comes from personal experience.
I am suffering since many years from a mild form of Ankylosing Spondylitis. Mostly non-spinal enthesitis. The condition is well controlled with an anti-TNFa antibody. Due to my travel constraints I am treated every six month, although every 4 month would be more ideal. It would be more ideal because usually after about 4-5 month the pain returns and I am struggling for a month and a half before I finally receive the next treatment. Since several years I endure this pain period. However, a couple month ago I made an astonishing discovery in a self-experiment. This discovery improved my situation immensely.
I subscribe to several ScienceDaily RSS feeds. Therefore I receive daily references to scientific publications that they feel relevant. A couple month ago I found in my RSS reader inbox a publication by a Japanese group that tested the efficacy of three known plant-based anti-inflammatory compounds in combination. It turned out that the combination of menthol (ME) with capsaicin (CA) significantly increased the potency of the anti-inflammatory effect, compared to each individual compound alone.
https://www.sciencedaily.com/releases/2026/04/260408225950.htm
https://www.mdpi.com/2072-6643/18/3/376
Here is a quote from the article’s discussion section: “We demonstrated that specific phytochemical combinations produce pronounced synergistic anti-inflammatory effects, most notably a ~700-fold reduction in the EC50 value for TNF expression upon co-treatment of CA with ME.
At the time I read this article my last anti-TNFa treatment was 4.5 month ago and I was already in pain. For that reason I thought I will give this a try. I had cayenne pepper in my pantry and I bought concentrated food grade menthol in a pharmacy. Based on what I thought would be reasonable I prepared a water/menthol/cayenne pepper/chocolate drink. I didn’t expect much but was really surprised by the effect. Right after the first time I consumed this drink the pain was completely gone and the effect lasted more than 12 hours.
Over the subsequent days I plaid around with the recipe, the amount of CA and ME, the frequency and the time points of ingestion. I settled on a delicious slightly spicy recipe once a day before bed time. Now I have an effective treatment that is capable of bridging the time from when the pain reappears to the time that I receive my next anti-TNFa antibody treatment.
I told my doctor about it. He is a MD/PhD and head of a university hospital rheumatology devision. He said, he will think about how to use this result in his medical practice.
What is the take-home message from this experience? Possibly there are many other plant- and microbial-based drug combinations out there which can potentially increase the efficacy substantial compared to the individual drugs alone. However, such increases in potency can only be identified with cell-based assays, animal experiments and clinical trials. Many such combination might be known already e.g. in Chinese medicine, however, many may not be known yet. Fortunately the medicinal properties of many plant and microbial compounds and their effect on particular molecular pathways in humans are known, which allows for the educated and systematic testing of such combinations. I think this might be a fruitful area of research.
I personally would experiment with such plant-based drug combinations on myself only after some data has been published. The chance is high that a particular combination has no additive or potentiating effect.
And basing body feeling and sleep quality on eating habits can be very subjective. Finding true cause-effect relationships will be difficult. However, some might be easy to find e.g. that eating too much garlic for dinner can cause restlessness resulting in a bad night of sleep.
Another problematic is that the number of biological medicinal compounds that one can add to one’s diet is limited. Essentially most compounds in natural products databases are derived from bacteria or fungal species. You cannot add these to your diet. In many other cases the amount of active compound found in a particular plant is too low to have any significant effect in a disease situation. And increasing the dose is often not reasonable or feasible. For example, compared to my anti-inflammatory recipe above, neither taking 100 fold more menthol or capsaicin as a single drug treatment is feasible. It would taste horrible and ruin your stomach. Therefore, finding more potentiating combinations via experimentation will make a big difference in my opinion. In addition to finding 3D analogs, predicting which combinations are worthwhile to experimentally test, might be a good application for an AI.
Here is another example of a natural compound that unfortunately is not found in high enough concentration in food products. NMN (Nicotinamide Mononucleotide) is a compound that is currently being studied for its anti-aging properties. NMN is a precursor used in the body for e.g. cellular energy production, DNA repair, and metabolism. The production of this compound in the body declines with age. Supplementation with NMN has been found to be beneficial, although results are still inconclusive.
My father is now 88 years old and in a phase of decline, although still relatively fit for his age. I suggested to him to add NMN to his Dietary Supplements a couple years ago. Since then I frequently have asked him whether he feels any improvement in his well being. He didn’t know. So, I suggested to him to stop taking NMN for a while to see what that would do. Yesterday he calls me and said I should buy some more NMN for him. His supply ran out 3 weeks ago. He said, his legs are now heavier, he is more tired during the day, his sleep got worse and his brain is foggy. He has been taking 300 mg NMN per day over the last couple years. It seems NMN did make a significant difference for him. It definitely makes sense for him to continue taking it.
Unfortunately, the amount of NMN in food products is low. Avocado is one of the food items with the highest concentration of NMN. By eating one average avocado one gets about 3 mg of NMN. My father would have to eat 100 avocados daily to get the 300 mg dose that he has been taking daily over the last couple years. That is not feasible. Therefore purchasing pure NMN gram wise is the only solution. Fortunately many vendors are in this market, unfortunately including many fraudsters.
I wonder whether alternative NMN-like cell energy boosters can be found in the FooDB database. I kind of doubt it, because NMN is not a drug molecule that could be substituted with a molecule of similar 3D structure. NMN is a metabolite that itself is consumed in the process. That is not something that a similar structured molecule can replicate. However, plant-based enzyme binders that boost or inhibit key enzymatic activity of enzymes in the NAD+ pathway may do the trick, if they exist.
CD38 has been found as a key negative regulator of NAD. CD38 increases during aging. And a small molecule inhibitor for CD38 has been shown to “Ameliorate Age-Related Metabolic Dysfunction by Reversing Tissue NAD+ Decline”.
https://pmc.ncbi.nlm.nih.gov/articles/PMC4911708/
https://www.cell.com/cell-metabolism/fulltext/s1550-4131(18)30194-3
https://www.nature.com/articles/s41574-025-01211-y
If a 3D analog to this small molecule CD38 inhibitor exits in the FooDB perhaps a food product could be identified that when eating with feasible amounts of NMN containing food products would be as effective as a 300 mg daily dose of NMN. It’s worthwhile to investigate it.